QMSR, Medical Devices
The 2026 QMSR Deadline: Why Medical Device Leaders Must Act Now
Alexander Ginzburg, Shahid Shah
December 2025
10 min read
Executive Summary
- On February 2, 2026, the FDA’s new Quality Management System Regulation (QMSR) will take effect, which aligns U.S. requirements with ISO 13485:2016 international standard.
- The FDA will conduct audits and inspections under the new QMSR framework. Holding an ISO 13485 certification does not exempt medical device manufacturers from FDA inspection requirements.
- With the 2026 deadline quickly approaching, device manufacturers have limited time to complete gap assessments, update their quality system documentation, and modernize supporting tools to ensure full readiness for the transition.
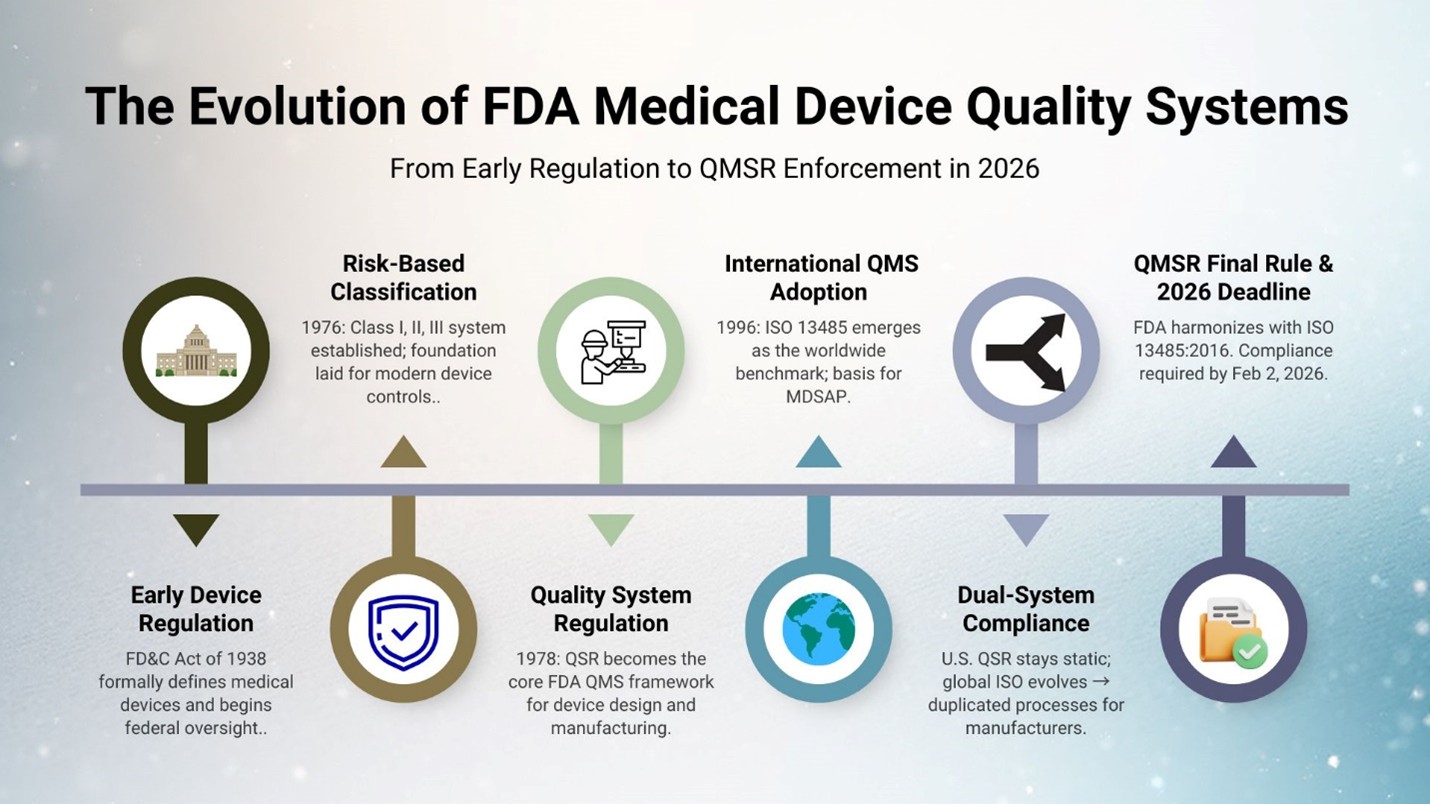
A historic change will soon take effect in how the FDA regulates quality management for medical device manufacturers. Beginning February 2nd, 2026, all medical device companies must be prepared for FDA governance and enforcement activities under the new Quality Management System Regulation (QMSR), which replaces the existing Quality System Regulation (QSR).
The FDA’s adoption of QMSR, harmonized with ISO 13485:2016, marks the most significant modernization of U.S. medical device regulation in decades. This transition is more than an FDA update or a “new version” as it represents a strategic shift toward alignment with global markets, requiring system-wide, risk-based quality management and readiness for broader FDA inspection scrutiny.
Here’s what leaders need to know about the scope and implication of the new QMSR regulation:
Who’s Affected:
- All medical device manufacturers subject to Part 820, including both U.S. and international companies whose devices are sold into the U.S.
- All third-party providers of services, materials, parts to the manufacturers of the finished devices
- All relevant functions within the medical device companies. The transition requires coordinated changes across product development, manufacturing, supplier oversight, regulatory, and enterprise governance functions. QMSR is not a Quality-only initiative.
- Indirect impact on venture capital companies, business development, M&A teams in MedTech companies due to the business risks. QMSR introduces material business risk.
What’s at Stake Before February 2026:
Medical device manufacturers have only a few months to the deadline to align their internal QMS policies, processes, procedures, documentation, or risk FDA inspection findings, including nonconformances, warning letters, release delays, and consequently loss of business. Under the previous QSR, some internal records (management reviews, internal audit records, supplier audits) were exempt from FDA inspection. However, QMSR grants FDA access to internal audits, supplier audit records, and management review records. For organizations navigating this transition, early gap visibility and inspection-oriented preparation are critical to avoiding downstream enforcement risk. Opsfolio works with medical device teams to assess QMSR readiness and identify inspection-visible gaps before enforcement begins.
What’s Changing: from QSR to QMSR
For decades, U.S. medical device oversight evolved in response to safety concerns and market inconsistencies from the 1938 FD&C Act’s first device definitions to the 1976 Medical Device Amendments that established today’s risk-based classification system. Meanwhile, ISO 13485 emerged as the global benchmark for medical device quality. As global supply chains, outsourced manufacturing, and multi-market approvals became the norm, the U.S. QSR no longer fully reflected how modern medical device organizations operate, increasing momentum for alignment with international standards. The FDA answered in 2024 with the new Quality Management System Regulation (QMSR), formally aligning U.S. requirements with ISO 13485:2016 and ISO 9000:2015, and signaling an ongoing commitment to global regulatory convergence and modernization.
The Final Rule can be found here: U.S. Food and Drug Administration
What Is Not Changing: FDA Inspection Authority
There will be a new inspection process replacing the existing Quality System Inspection Technique (QSIT). Although there is an established certification process through independent certification bodies for ISO 13485, and device manufacturers may choose to obtain it for international conformance, the FDA will neither issue nor accept such certifications in lieu of an FDA inspection. Do not assume that certification to ISO 13485 equates to full FDA compliance.
From QSR to QMSR: Structural and Technical Changes
The FDA’s shift from the long-standing QSR to the new QMSR, harmonizes U.S. device quality requirements with ISO 13485:2016 and ISO 9000:2015 terminology and requirements. While the foundations for key areas remain intact, manufacturers must adopt ISO 13485 language, strengthen risk-based thinking, and expand integration of risk management throughout the product lifecycle via ISO 14971 principles. Expectations for record management are also raised, requiring manufacturers to shift Device Master Records and Device History Records toward ISO-style documentation structures and reinforcing labeling controls.
1. Terminology and Design Controls
The shift from the QSR legacy terminology to ISO-aligned terminology means companies must update all QMS-managed documents to avoid discrepancies and inconsistent use of terms across U.S. and international markets. In an effort to align the vocabularies and avoid confusion, the FDA has eliminated conflicting terminology to ensure that the QMSR requirements are clear to device manufacturers and leave no room for misinterpretation. With that, it is important to note that several existing definitions, including such familiar terms as “manufacturer,” “device,” and “product” (which may also mean “service”) will supersede the corresponding definitions in ISO 13485.
Here is a crosswalk of a few terms from both systems which are key to design controls and engineering:
| QSR Terms | QMSR / ISO Terms | ISO 13485 Clause | Notes |
|---|---|---|---|
| Design History File (DHF) | Design & Development File (DDF) | 7.3.10 | Map DHF to DDF and preserve the history of changes |
| Device Master Record (DMR) | Medical Device File (MDF) | 4.2.3 & 7.5 | Broader term to include the information from both DMR and DHR |
| Device History Record (DHR) | Medical Device File (MDF) Production / Service Records | 7.5 |
In many cases, the changes are mostly semantic or terminological.
Importantly, QMSR does not require manufacturers to recreate historical design documentation. Because core design control processes remain substantially similar, existing Design History File (DHF) content can typically be reused provided it is correctly mapped, structured, and aligned to ISO 13485 terminology within the new Design and Development File.
2. Labeling and Packaging
QMSR elevates labeling and packaging from a supporting compliance function to a clearly defined, inspection-visible control area. While labeling requirements existed under the prior framework, QMSR increases the likelihood that labeling, storage, and handling deficiencies surface as inspection findings — raising the operational and regulatory impact of mislabeling errors.
One of the newly added section (Section 820.45) specifically deals with device labeling and packaging controls. It does not replace the existing 21 CFR Part 801 - Labeling but rather strengthens and augments it.
The Section 820.45 asks that the label and/or packaging must contain an accurate:
- UDI (An internationally recognized Unique Device Identifier)
- Expiration date
- Storage instructions
- Handling instructions
- Processing instructions
3. Risk Management
In QMSR, risk management becomes system-wide and embedded into all processes, not limited to design controls. The FDA now expects application of risk-based decision-making practices across the entire QMS, including suppliers, records, labeling, and post-market activities. For many medical device manufacturers, this represents the most challenging shift. While risk management practices often exist in pockets, QMSR requires evidence that risk-based thinking is consistently applied, documented, and monitored across functions.
Transitioning from QMS to QMSR must result in a revised risk management approach based on collected evidence and risk management techniques such as risk identification, logging, categorization, and mitigation. This must be incorporated into every major QMS process across the management system. (supplier management, manufacturing, service, records, labeling, etc.).
While in some areas risk is explicitly defined as a requirement: for example, ISO 13485, Clause 7.3(g) on Design Validation other sections carry an implicit expectation to incorporate risk management practices. QMSR enforcement extends risk-based expectations across the entire quality system
Where most organizations will face gaps is not in documented procedures, but in how risk is operationalized and evidenced across the business. Under QMSR, FDA scrutiny extends beyond design controls into supplier oversight, labeling, records, and post-market activities, requiring documented risk-based decisions, ongoing monitoring, and lifecycle risk management. Previously lower-visibility areas such as internal audits, management reviews, and supplier performance records are now squarely within inspection scope. Companies must also align terminology and documentation structures with ISO 13485 expectations and demonstrate, through accessible records, how risk-based thinking is consistently applied across all QMS processes.
How QMSR Expands Risk Expectations Beyond QSR
| Domain/Area | QMS | QMSR | Comments |
|---|---|---|---|
| Scope | Mostly Design Controls | Entire QMS + Product/Solution lifecycle + regulatory risk | Expand risk management beyond design |
| Decision Making | Informal | Formal documented risk decisions, controls scaled by risk, monitoring/trending | Update procedures to reflect “risk-based decision” |
| Supplier controls | Supplier evaluation mostly | Supplier evaluation + ongoing performance monitoring + risk-based supplier controls | Add supplier monitoring metrics and risk criteria |
| Records and Audits | Internal audits and reviews may be less visible | Internal audits/management reviews/supplier audits are clearly within FDA inspection scope | Ensure records are accessible, documented, traceable |
| Labeling/Packaging | Covered but less prescriptive | Clause §820.45 controls for labeling/packaging, storage/handling, risk of mis-labeling | Map labeling/packaging risk and controls explicitly |
| Regulatory/lifecycle risk | Possibly separated | Integrated within QMS: design → manufacture → post-market → regulatory compliance | Expand lifecycle risk management, integrate regulatory risk |
| Terminology and Alignment | Use of deprecated terms DMR/DHR/DHF | Terminology aligned with ISO 13485; legacy terms phased out; FDA definitions override whenever there is a conflict | Update documents/records to align with new terms |
Opsfolio supports organizations in translating risk requirements into inspection-ready evidence across suppliers, records, labeling, and post-market processes.
Three Strategic Priorities for a Successful Shift to ISO 13485 Alignment
For medical device manufacturers operating legacy QMS frameworks based on the QSR, the shift requires investment in risk management, documentation structure, supplier oversight, and the quality system itself. The transition elevates expectations around building a medical device product or service, emphasizing risk-based approaches and evidence-based quality management. This transition also presents an opportunity to mature quality management systems by incorporating emerging AI-based automation tools to strengthen integration across design, production, post-market surveillance, cybersecurity, and service/complaint handling.
While the transition may feel substantial, the path forward becomes clear when the work is focused on three strategic priorities.
1. Modernize and Align Your QMS
The first step is ensuring your QMS aligns with ISO 13485:2016. This starts with a comprehensive gap assessment to identify where existing QSR-based processes diverge from QMSR expectations. Manufacturers should update documentation, terminology, and tooling, specifically design controls, supplier controls, and change management to the modernized ISO framework. Supporting software and process automation tools must also be validated to ensure they can reliably meet QMSR requirements. This modernization effort lays the foundation for a compliant, efficient, and audit-ready QMS.
2. Elevate Risk, Quality, and Inspection Readiness
Risk management is one of the most significant areas of change as QMSR fully embraces ISO 14971 principles. Companies should strengthen risk processes across the entire lifecycle from design to production to post-market, ensuring traceability and integration into decision-making. With FDA inspections expected to shift to an ISO-aligned approach, teams must refresh internal audits, update product release practices, and run mock inspections to build confidence and readiness. This is where organizations will feel the operational impact of the new regulation most clearly.
3. Drive Organizational Adoption, Governance, and Communication
Regulatory change succeeds only when the organization moves together. That means establishing clear governance, a transition roadmap, and cross-functional accountability. Training is essential, not just for Quality teams, but for R&D, manufacturing, supply chain, and executive leaders who must understand their role in sustaining compliance. A well-structured communication plan ensures transparency, alignment, and consistent messaging so the entire company progresses toward QMSR compliance with clarity and purpose.
Next Step:
Opsfolio’s QMSR Readiness Assessment helps medical device organizations identify inspection-visible gaps, align documentation and risk practices with ISO 13485, and prepare for ISO-aligned FDA inspections ahead of the February 2026 deadline.
The Bottom Line:
QMSR represents a historic shift toward global alignment, which comes with a higher bar for documentation, risk management, and inspection readiness. Organizations that modernize now will reduce audit exposure, streamline quality operations, and strengthen their ability to scale in global markets. Opsfolio’s CaaS platform can help accelerate the transition with structured assessments, automated documentation workflows, and end-to-end readiness support.